Qu’est-ce que le syndrome de Dravet ?
Le syndrome de Dravet est une maladie génétique neurologique rare caractérisée par une épilepsie sévère associée à des troubles du développement.
Estimation d'occurence :
Entre une naissance sur 20 000 et une naissance sur 40 000
Elle est le plus souvent liée à une mutation génétique dite « de novo », ce qui veut dire que la mutation apparait alors que les parents ne la possèdent pas dans leur patrimoine génétique.
L’épilepsie débute avant l’âge d’un an par des crises convulsives souvent déclenchées par de la fièvre. Les premières crises d’épilepsies sont souvent des crises longues, des états de mal épileptiques. Elles se manifestent par des secousses musculaires avec une perte de connaissance. Par la suite, les enfants ont des crises d’épilepsie de divers types. Malgré les traitements, les personnes atteintes du syndrome de Dravet continuent généralement de faire des crises tout au long de leur vie.
Ce syndrome entraîne un retard de développement souvent évident après l’âge de 3-4 ans. Les enfants peuvent avoir des difficultés de langage, une mauvaise coordination des mouvements, une déficience intellectuelle, des troubles autistiques, des troubles du comportement, des troubles du sommeil et de l’alimentation et des difficultés motrices.
Chaque enfant ou adulte atteint du syndrome de Dravet est différent. L'expression des symptômes est variable selon les personnes.
On ne sait pas exactement combien de personnes en France sont diagnostiquées. On estime qu’elles sont entre 600 et 800.
La maladie a été décrite en 1978 par le docteur Charlotte Dravet. Le lien avec une mutation du gène SCN1A a été décrit en 2000.


L'évolution du syndrome de Dravet
Premières manifestations
La première manifestation de la maladie est une crise d’épilepsie qui a lieu généralement entre les 3 mois et les 1 an de l’enfant.
C’est, dans la plus part des cas, une crise clonique (des convulsions) ou hémiclonique (des convulsions d’un seul côté du corps) plutôt longue, qui peut être un état de mal épileptique (crise d’épilepsie de plus de 30 minutes). Elle se manifeste généralement dans un contexte fébrile consécutif à une infection ou à un vaccin, ou d’élévation de température (chaleur, bain chaud).
Elle peut conduire les bébés à être hospitalisés en soins intensifs et à passer quelques jours à l’hôpital. Elle passe souvent pour une crise fébrile particulièrement longue.
À ce moment de la maladie, les examens médicaux qui sont faits (IRM, EEG… ) sont généralement normaux.
Quelques jours ou quelques semaines après le premier épisode les crises réapparaissent et se répètent.
« La première crise … C’était un jour comme un autre, une visite de routine chez le pédiatre pour les premiers vaccins. C’était un retour à la maison, une fin d’après-midi et là, notre bébé qui a des secousses de la tête et qui nous regarde avec un regard vide, puis c’est au tour de la tête et d’un bras et puis l’autre et puis tout le corps qui se met à secouer. C’étaient les secours qui mettent toujours trop longtemps à arriver. Puis, après une éternité à tenter de faire cesser ces secousses, c’est enfin l’apaisement une respiration normale un long sommeil et le début d’une longue série de ce jour qu’on n’aurait jamais voulu vivre. » Marco, papa de Maxime
Vers un diagnostic
C’est la répétition des crises, leur type et leur durée qui mettent les neuropédiatres sur la piste du syndrome de Dravet. Le diagnostic est important, car certains médicaments antiépileptiques sont contre-indiqués et il existe des traitements spécifiques pour le syndrome de Dravet (Les Traitements).
Le diagnostic est un diagnostic clinique (c’est-à-dire basé sur les observations des symptômes que font les neuropédiatres). Mais comme dans 85% des cas environ la maladie est due à une mutation du gène SCN1A, les spécialistes proposent de faire une analyse génétique.
Quand le médecin pose le diagnostic, il met en place des traitements adaptés à la maladie. Ces traitements seront surement réévalués en fonction de leur efficacité et des effets secondaires observés.
Cependant, le syndrome de Dravet reste une maladie rare. Il est parfois mal connu des pédiatres et neuropédiatres. Il peut être utile de solliciter un centre de référence épilepsies rares (Centres de référence) pour avoir l’avis d’un spécialiste.
Souvent les résultats de l’analyse génétique arrivent plus tard (ça peut être long…). Les résultats peuvent confirmer le diagnostic en mettant en évidence une mutation du gène SCN1A.
Si la mutation SCN1A n’est pas retrouvée, il se peut qu’elle n’ait pas été détectée lors de l’analyse génétique. Il est aussi possible qu’il n’y ait pas de mutation de SCN1A, mais que ce soit d’autres gènes qui soient impliqués. Cela ne modifie cependant pas le diagnostic clinique de syndrome de Dravet.
« Le diagnostic : sans lui on est perdu, complètement sans repère. En même temps se dire qu’il pourrait être confirmé alors que Google vous a déjà renseigné de manière brutale sur le syndrome suspecté, c’est affreux. Tant que le verdict n’est pas tombé, on espère qu’il s’agit de quelque chose de moins grave. Assez humain comme espérance…
Il m’a fallu des mois pour « digérer » la nouvelle, surtout car plusieurs neurologues n’avaient pas pu s’empêcher de dire « non, il ne faut pas que cela soit le Syndrome de Dravet ». Pas trop rassurant (c’est un euphémisme).
Finalement c’est une généticienne appelée pour savoir si nos résultats des tests génétiques ne s’étaient pas perdus qui m’a expliqué, dans un langage de science-fiction pour moi, non scientifique, que la mutation était bien celle du SD. Soulagement et effondrement en même temps » Fabienne, maman de Tom, diagnostiqué en 2008
Évolution dans l'enfance
Avec la mise en place des traitements et l’évolution de la maladie, les crises deviennent généralement moins longues, mais plus fréquentes. D’autres types de crises peuvent aussi apparaitre : des crises myocloniques, des absences atypiques, des crises focales. (Crises d’épilepsie)
Les traitements permettent de diminuer le nombre de crises et leur durée, mais, la plupart du temps, les personnes atteintes du syndrome de Dravet continuent de faire des crises tout au long de leur vie.
À côté de cette épilepsie, le retard de développement et les troubles associés deviennent souvent évidents entre 2 et 4 ans. Dans l’enfance, les personnes atteintes du syndrome de Dravet présentent souvent des difficultés cognitives et d’apprentissage. La différence avec les enfants du même âge va s’accentuer au cours du temps. (Troubles associés)
« Après plus de 500 crises en 7 ans, nous avons bien évolué et appris à gérer. L’épilepsie est presque passée au second plan, aujourd’hui, on fait avec. Et malgré les risques, on essaie de vivre, de tenter des trucs. Parfois, ça ne passe pas, mais de temps en temps on a de bonnes surprises, et ces petites victoires ont beaucoup plus de saveurs que chez les autres » Maher, papa de Gabriel
Evolution à l’adolescence et à l’âge adulte
Il est important de préciser que le syndrome de Dravet entraine des conséquences très variables d’une personne à l’autre. Les comparaisons et les projections d’évolution sont donc très difficiles à faire. Nous avons essayé ici d’illustrer des difficultés typiques. Cela ne signifie pas qu’elles concernent toutes tous les dravets !
A l’adolescence et à l’âge adulte, l’épilepsie passe souvent au second plan. Même si la poussée de croissance et la puberté peuvent conduire à une augmentation des crises d’épilepsie qui entraine des ajustements de traitements, pour certaines personnes les crises peuvent disparaitre pendant de longues périodes à l’âge adulte.
Des troubles moteurs peuvent se développer chez les personnes atteintes du syndrome de Dravet, il s’agit le plus souvent d’une ataxie qui provoque une posture chancelante et une démarche instable, de tremblements, et parfois d’un trouble de la démarche appelé crouch gait (démarche semi-accroupie). Ces difficultés motrices limitent la marche longue et certaines personnes ont besoin d’un fauteuil roulant.
Les difficultés cognitives, les troubles du comportement et les troubles neurodéveloppementaux ont également un impact important sur le quotidien.
Les adultes atteints du syndrome de Dravet sont généralement partiellement dépendants ou vivent dans des institutions spécialisées.
Diagnostic tardif
De plus en plus d’adultes sont diagnostiqué tardivement. Lorsqu’ils étaient enfants, le syndrome de Dravet était moins connu et n’était pas aussi bien diagnostiqué qu’aujourd’hui. Ce diagnostic est important car il permet, d’éviter les traitements proscrits, de mettre en place des traitements spécifiques, de mieux comprendre certains troubles associés et il permet à leurs proches de rencontrer des personnes qui ont le même vécu qu’eux.
Risque de mortalité
Le risque de décès lié au syndrome de Dravet est réel, même s’il est globalement faible. Plus de 85% des personnes touchées grandissent et deviennent adulte.
Les décès prématurés peuvent être dus :
- à un accident lors d’une crise (chute, noyade)
- à une encéphalopathie aiguë lors d’un état de mal
- ou à une SUDEP.
On parle de SUDEP (Sudden Unexpected Death in Epilepsy ou mort subite inattendue en épilepsie), lorsqu’une personne épileptique décède et qu’on ne trouve pas la cause de son décès. Les médecins et les chercheurs n’en connaissent pas précisément les causes ou l’origine. Dans le cadre d’un syndrome de Dravet avec mutation du gène SCN1A, on sait que les canaux sodiques ont aussi un rôle dans le fonctionnement cardiaque, mais on ne sait pas si ça peut être une cause de SUDEP.
Même si on ne peut rien faire quand une SUDEP à lieu, les travaux de recherches en cours laissent penser que le risque est réduit par un traitement bien adapté. C’est donc en travaillant de concert avec le neurologue à la recherche d’un traitement optimal pour diminuer au maximum le nombre de crise tout en minimisant les effets secondaires des traitements que l’on pourra essayer de limiter les risques.
D’autre part, vous pouvez envisager l’utilisation un détecteur de crise et d’un un oreiller anti-étouffement.
Il est aussi recommandé de rester près de la personne dans les minutes suivant la fin d’une crise d’épilepsie.
L’Alliance Syndrome de Dravet est présente auprès des familles ayant perdu un proche atteint du syndrome de Dravet. (Deuil)
« Nous vivons au jour le jour et sans trop se projeter. Nos enfants nous font voir la vie différemment, on profite de chaque instant. Savoir que demain cela peut arriver chez nous aussi, nous embrassons chaque soir notre enfant en espérant très fort à demain matin » Anne-Sophie, maman de Fleur
« Nous avons perdu nos deux petits jumeaux de SUDEP. Ils avaient 3 ans et presque 6 ans. Il n’y a pas de mots pour dire ce qu’on a vécu. Je peux juste remercier les personnes qui nous en avaient parlé. On n’est jamais prêt à « ça », mais avoir su que c’était une éventualité nous a aidé. » Manue, maman de Joseph et Antoine
Génétique du syndrome de Dravet
Le gène SCN1A est un gène du chromosome 2. Comme pour tous les gènes, nous en possédons deux exemplaires. Ce gène code pour la protéine NaV1.1 : la sous unité alpha du canal sodique, voltage dépendant, type 1. Cette protéine est une partie des canaux sodiques, qui permettent de faire passer des ions sodium au travers de la membrane cellulaire (la paroi de la cellule). Ils jouent un rôle important dans l’excitabilité des neurones.
Dans le cadre du syndrome de Dravet, la mutation d’un des deux gènes SCN1A entraine une « perte de fonction » des canaux sodiques, c’est-à-dire qu’ils ne font pas passer autant d’ions sodium qu’ils devraient le faire. Les protéines fabriquées par le gène qui n’est pas muté ne sont pas suffisantes pour faire fonctionner normalement les cellules.
Dans le syndrome de Dravet la mutation est généralement « de novo » ce qui veut dire qu’elle est présente alors que les parents ne la possèdent pas dans leur patrimoine génétique. Dans de rares cas, la mutation est héritée d’un des parents, chez qui elle ne s’exprime pas ou peu. L’analyse génétique permet de le déterminer. Une personne porteuse du gène muté a un risque de 50% de transmettre la mutation.
Si mon proche a une mutation du gène SCN1A, est ce qu’il a le syndrome de Dravet ?
Une mutation du gène SCN1A peut entrainer un syndrome de Dravet, mais pas forcément…
Voici le spectre des conséquences d’une mutation du gène SCN1A :
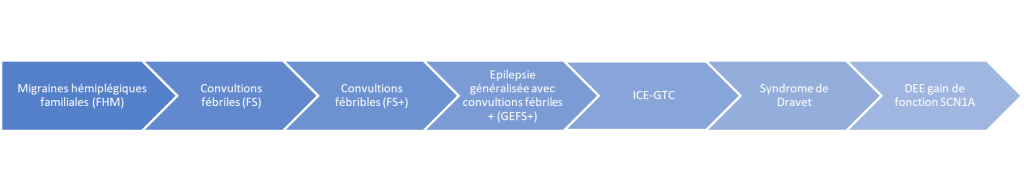
C’est la clinique (âge de la première crise, fréquence, type de crises, développement…) qui permettent au spécialiste de faire le diagnostic de syndrome de Dravet.
Et si l’analyse génétique de mon proche n’a pas trouvé de mutation du gène SCN1A ?
Dans ce cas, soit la mutation du gène SCN1A n’a pas été trouvée lors de l’analyse (ce qu’il ne veut pas dire qu’elle n’existe pas), soit il n’y a pas de mutation du gène SCN1A. Une mutation d’un autre gène peut aussi être trouvée.
On n’a pas trouvé de mutation du gène SCN1A chez 15% environ des personnes atteintes du syndrome de Dravet. Si le tableau clinique correspond, votre proche peut être atteint du syndrome de Dravet sans qu’il y ait de mutation génétique SCN1A connue.

Fiche Orphanet
Accueil Orphanet est une ressource rassemblant des informations sur les maladies rares à destination des médecins et du grand public. ...

20 questions sur le SD
Accueil Consulter le PDF Voici le guide : « les 20 premières questions sur le syndrome de Dravet. » Les auteurs sont des ...